
Use my own data in Ensembl
NGS reads and more
Ensembl supports a number of different filetypes for upload and visualisation along the genome.
The most popular page in which to view your data is the Location tab, Region in Detail. Use the 'Custom tracks' button in the left hand menu.
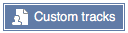
Select a file on your computer, or paste in a URL or a block of data, and a dropdown list will appear showing available formats. If your file has an unambiguous extension, e.g. '.bam', the format will be preselected, otherwise you will need to select it yourself.
We upload plain text files such as BED or GFF (up to a limit of 20MB); large indexed formats such as bigBed and BAM can only be attached via a URL.
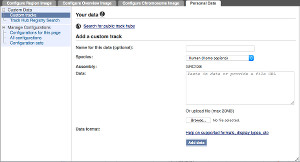
Here is an example of sequence reads attached as a BAM file, along chromosome 20.

Find out more:
Alleles and mutations
Use the Variant Effect Predictor (VEP) to determine effects of nucleotide substitutions, insertions and deletions within genes or the genome.
Genes and Transcripts
Use BioMart to get information about a list of gene or transcript IDs, names, symbols, or chromosomal locations. Some examples:
- Convert Ensembl IDs to gene names
- Get SNPs for a list of transcripts
- Export protein sequences for genes on a chromosomal region
Learn how to use it in this 5 minute video:
BLAST
If you have sequence, consider BLAT (the Blast Like Alignment Tool) or BLAST against Ensembl genes, genomes, cDNA or protein.

For more about BLAT/BLAST read the help page.
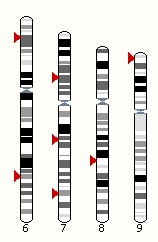
Genome-wide data
Your data can be displayed on Ensembl karyotypes, by adding your data and going to the 'Whole Genome' view. Note that sparse data can be displayed as points, whereas larger datasets can only be displayed as density tracks.
You can also create a karyotype display showing the location of selected Ensembl features by going to Location/Genome and clicking on 'Add features'.
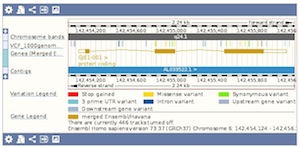
You can display your own genome-wide variation data in Ensembl by attaching your data as VCF files. See our FAQ for more details. Visualise different consequence terms of your variants on the set of Ensembl genes and transcripts. The different consequence terms are displayed by different colours in the Region in Detail view.
![[Click to enlarge]](/img/custom_data_2.jpg){kind=link}
![[Click to enlarge]](/img/custom_data_3.jpg){kind=link}
![[Click to enlarge]](/img/custom_data_4.jpg){kind=link}
![[Click to enlarge]](/img/custom_data_7.jpg){kind=link}